Understanding Single-Cell RNA Sequencing (scRNA-seq)
Single-cell RNA sequencing (scRNA-seq) is a cutting-edge technology that allows scientists to examine the gene expression profiles of individual cells within a tissue. Unlike traditional methods that analyze gene expression in bulk cell populations, scRNA-seq provides a detailed view of the biological heterogeneity, or differences, among various cell types in a tissue. This detailed insight is crucial for understanding how different cells function and interact within their environment.
The Challenge: Inferring Cell Types
A major task in scRNA-seq analysis is determining the types of cells present in a tissue sample. This is essential for downstream research, such as identifying disease mechanisms or discovering new therapeutic targets. Traditional methods for cell type inference often focus on genes that are highly variable and have high expression levels. However, this approach overlooks genes that, despite their lower expression levels, can still provide valuable information about cell identity.
Introducing scLEGA: A Novel Approach
To address this limitation, researchers at Northeast Forestry University have developed a new method called scLEGA. This innovative approach aims to improve the accuracy of cell type inference by considering genes with both high and low expression levels. Here’s how scLEGA works:
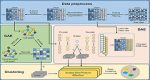
Zero-Inflated Negative Binomial (ZINB) Loss Function: scLEGA uses a unique mathematical function called the ZINB loss function. This function helps in accurately modeling the contribution of genes with lower expression levels, which are often ignored by traditional methods.
Multi-Head Attention Mechanism: scLEGA combines two distinct clustering strategies using a multi-head attention mechanism. This advanced computational technique allows the method to focus on different parts of the data simultaneously, improving the overall clustering accuracy.
Low-Expression Optimized Denoising Autoencoder: To handle the noise and missing data commonly found in scRNA-seq datasets, scLEGA uses a denoising autoencoder. This is a type of artificial neural network that helps clean the data and reduce its dimensionality, making it easier to analyze.
Graph Autoencoder (GAE): scLEGA also includes a graph autoencoder that uses information from neighboring cells to guide the dimensionality reduction process. This helps in capturing the relationships between cells more accurately.
Iterative Fusion of Denoising and Topological Embedding: The method iteratively combines the cleaned data from the denoising autoencoder with the structural information from the graph autoencoder. This fusion creates a hidden representation of the data where similar cells are clustered closer together.
The overall structure of scLEGA
scLEGA, through the implementation of changed ZINB loss, is able to focus on the contributions of genes with lower expression levels to cell type inference. This loss function is integrated into a DAE to learn denoising embedding. Subsequently, multi-head attention is employed to combine the denoising embedding produced by the DAE with the topological embedding generated by the GAE for deep clustering purposes.
Superior Performance
When compared to 12 other state-of-the-art methods on 15 different scRNA-seq datasets, scLEGA showed superior performance in several areas:
Clustering Accuracy: scLEGA provides more accurate clustering of cells, which is crucial for correctly identifying cell types.
Scalability: The method is efficient and can handle large datasets, making it suitable for modern high-throughput scRNA-seq experiments.
Stability: scLEGA produces consistent results, which is important for reliable scientific research.
Conclusion
Single-cell RNA sequencing has revolutionized our ability to study the diversity of cells within tissues. With the introduction of scLEGA, researchers now have a powerful tool that improves cell type inference by considering both highly and lowly expressed genes. This advancement not only enhances the accuracy of cell type identification but also opens up new avenues for exploring the complexities of biological systems. As scRNA-seq technology continues to evolve, methods like scLEGA will be instrumental in uncovering the details of cellular functions and interactions.
Availability – The scLEGA model codes are freely available at https://github.com/Masonze/scLEGA-main.
Liu Z, Liang Y, Wang G, Zhang T. (2024) scLEGA: an attention-based deep clustering method with a tendency for low expression of genes on single-cell RNA-seq data. Brief Bioinform 25(5):bbae371. [article]