Single-cell RNA sequencing (scRNAseq) has transformed biology by allowing scientists to study the gene expression of individual cells. This technique provides deeper insights into how cells function, develop, and respond to diseases. However, as the use of scRNAseq grows and researchers analyze larger datasets, the need for powerful computing resources becomes critical. The complexity of analyzing this data often requires coding knowledge and high-performance computing (HPC) systems, which aren’t accessible to everyone.
The Challenge
The problem is twofold:
Many existing tools for analyzing scRNAseq data are either too simple or too complicated. Web apps that are easy to use can’t handle large datasets, while advanced tools that can manage complex analyses require significant coding skills.
With more data comes the need for more computational power. Local workstations (like regular desktop computers) often struggle to process large datasets, leaving many researchers searching for solutions that can keep up with the growing demands of scRNAseq studies.
Introducing scRNAbox
To address these challenges, a team of researchers at McGill University has developed scRNAbox, a pipeline specifically designed for HPC systems. Think of scRNAbox as a toolkit that helps researchers handle every step of the scRNAseq analysis process from start to finish—without the need for advanced programming knowledge.
Here’s how it works:
High-performance computing (HPC) systems: scRNAbox is built to run on powerful computing systems, ensuring that even large datasets can be processed quickly and efficiently.
SLURM workload manager: This is a system used by HPCs to manage tasks. scRNAbox uses it to handle multiple tasks at once, speeding up the process.
End-to-end analysis: scRNAbox covers every part of the scRNAseq process, from quality control (to ensure data accuracy) to clustering cells into groups, identifying cell types, and comparing gene expression between different conditions.
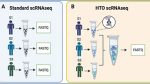
ScRNAbox analysis workflow
The scRNAbox pipeline provides two analysis tracks: 1) standard scRNAseq and 2) HTO scRNAseq. A Standard scRNAseq data is prepared by sequencing each sample separately, resulting in distinct FASTQ files for each sample. B HTO scRNAseq data is produced by tagging the cells from each sample with unique oligonucleotide “Hashtag” conjugated antibodies (HTO). Tagged cells from each sample are then pooled and sequenced together to produce a single FASTQ file. Sample-specific HTOs are used to computationally demultiplex samples downstream. C Steps of the scRNAbox pipeline workflow. Steps are designed to run sequentially and are submitted using the provided bash scripts through the command line. scRNAbox takes FASTQ files as input into Step 1; however, the pipeline can be initiated at any step which takes the users processed data as input
Key Features of scRNAbox
User-friendly: Researchers don’t need advanced coding skills to use it. Once set up, the pipeline does all the heavy lifting.
Handles large datasets: Unlike web apps that can only process smaller datasets, scRNAbox can handle large, complex datasets, making it ideal for researchers studying numerous cells at once.
Comprehensive analysis: scRNAbox goes beyond just processing data. It helps researchers group cells into clusters, identify the types of cells in the dataset, and compare how genes are expressed in different conditions or groups of cells.
Versatile: The pipeline can analyze both regular and “Hashtag” samples—where cells are tagged with unique identifiers to track them through the process.
Real-world Application
The researchers tested scRNAbox on two publicly available datasets, demonstrating its power and efficiency. By analyzing these datasets, scRNAbox showcased its ability to quickly and accurately process large amounts of scRNAseq data.
Conclusion
scRNAbox is an innovative tool for the field of single-cell RNA sequencing. It bridges the gap between the high computational demands of scRNAseq data and the accessibility of tools for non-expert users. With scRNAbox, researchers can process large datasets and gain valuable insights without the steep learning curve of coding. This new tool is a step forward in making advanced genomic analyses more accessible to the broader scientific community.
Availability – A complete user guide and the code used in this manuscript can be found at the scRNAbox GitHub site: https://neurobioinfo.github.io/scrnabox/.