When scientists study cells, they often focus on how genes are turned on and off, and how the instructions in our DNA are turned into proteins through a process involving RNA. New technologies, known as single-cell genomics, allow researchers to look at both DNA and RNA in a single cell at the same time, which provides a detailed picture of what’s happening inside. But this creates a challenge: how do you make sense of all that data when cells can be quite different from one another, and when processes like gene expression can vary so much?
What is Multimodal Single-Cell Genomics?
Multimodal single-cell genomics refers to the ability to measure different aspects of DNA and RNA processing within individual cells. Essentially, this means scientists can look at the activity of genes (through RNA) and changes in the genome (through DNA) in one go. By doing this, they can study complex biological processes in a way that wasn’t possible before.
For example, they can observe how different types of cells develop from stem cells (a process known as cell fate) or how cancer cells grow and spread by looking at how genes are spliced incorrectly. This is incredibly useful for studying diseases like cancer and for understanding how our bodies develop.
The Problem with Clustering Cells
One of the biggest challenges in this field is figuring out how to group cells based on the data collected, which is called “clustering.” By clustering, scientists can determine which cells are behaving similarly, which might indicate that they share the same type or function.
However, current methods for clustering cells often involve a lot of guesswork. Many approaches don’t fully account for the natural properties of the data or how DNA and RNA processes relate to each other. This can make the results less reliable.
meK-means: A New Way to Cluster Cells
Researchers at Caltech have developed a new method called meK-means (mechanistic K-means), which improves how we cluster cells by focusing on how DNA and RNA are physically related. Instead of just looking at gene activity and clustering cells based on similarities, meK-means takes a more sophisticated approach. It uses the fact that new RNA (nascent RNA) and mature RNA are connected in a physical, causal way within cells.
Here’s a simplified way to think about it:
Nascent RNA is like a draft of a message that the cell is writing.
Mature RNA is the final version of that message, ready to be sent out for protein production.
By clustering cells based on how these two types of RNA interact, meK-means helps researchers understand the deeper, shared processes happening across different cells. In other words, it looks at the machinery inside the cell that controls RNA production and uses that information to group the cells.
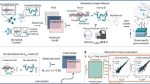
Mechanistic K-means inference and simulation performance
a, Diagram of data generation and meK-means inference. Input: multimodal sequencing data with underlying states Z. Output: inferred states Z^ with state-specific biophysical parameters. The meK-means model defines underlying states with governing rates describing transcription and sequencing. b, Parameters for gene markers, with increased burst size b or decreased splicing β/k, selected for KSim states. Counts sampled for each state to make input data X. i, Table of clustering methods tested with possible data input options. ii, ARI and AMI scores for each method, n = 3 runs, compared with true clusters, across possible inputs for the simulations (with 1, 5 or 10 simulated clusters). For methods with a K hyperparameter, the same K as the data was used; otherwise, the default Leiden resolution parameter was used. *P < 0.05 from a one-sided t-test (3.6 × 10–195 for KSim=5 and 8 × 10–57 for KSim=10). neg., negative. c, i (left): correspondence plot between meK-means-inferred clusters and true clusters for the negative control simulation. Values denote overlapping cell counts. i (right): correlation of the burst size and splicing rates across genes, to the true parameters, shown for the boxed cluster. ii, Same plots as c (i) shown for the KSim=5 simulation. For true cluster 2, the inferred parameters from the corresponding cluster (inferred cluster 0) are shown in 2D and 3D space (including degradation γ/k), with distributions for burst size and splicing rate of the markers. iii, Same plots as c (ii) for the KSim=10 simulation. Marker plots shown for inferred cluster 0 (true cluster 6). All parameter values are shown in log10; θ^ denotes the inferred parameter. The dashed lines are the identity line (y = x).
Why This is Important
With meK-means, researchers don’t have to make arbitrary choices about how to balance different kinds of data or how to cluster cells. Instead, they can rely on the physical processes happening inside the cells to guide them. This method makes it easier to understand what’s driving changes in cells, whether in healthy development or in diseases like cancer. It gives scientists a clearer picture of how cells function at a fundamental level.
A New Way to Define Cell Types
One of the key takeaways from meK-means is that it provides a new way to define what a “cell type” really is. Instead of just grouping cells based on what they look like under the microscope or based on simple measurements of gene activity, this method looks at the biophysical processes that drive cell behavior. This could lead to more accurate definitions of cell types and a better understanding of diseases.
In summary, meK-means is a powerful tool that helps scientists uncover the hidden mechanisms of cells by focusing on how they process RNA. It improves the way we group and study cells, leading to better insights into how our bodies work and how diseases develop.