The field of single-cell RNA sequencing (scRNA-seq) has grown rapidly, allowing researchers to explore gene expression at an unprecedented resolution. As the volume and complexity of this data increase, the need for robust methods to analyze and integrate it has become more pressing. One of the latest advancements in this area is a method known as scParser, which offers new ways to understand how different biological conditions affect various cell populations.
Addressing the Challenges of Data Integration
Single-cell RNA sequencing provides a detailed snapshot of gene expression in individual cells, helping scientists uncover the diverse roles that genes play in health and disease. However, integrating this vast amount of data to understand how different conditions influence cell behavior can be challenging. Existing methods often fall short in capturing the nuanced effects of these conditions across various cell types.
Introducing scParser: A Scalable Solution
To tackle this challenge, researchers at the Chinese University of Hong Kong have developed scParser, a scalable approach designed to model and analyze the diverse effects of biological conditions on cell populations. Unlike previous methods, scParser focuses on understanding how gene expression changes contribute to different phenotypes by examining the specific impacts on subpopulations of cells. This approach helps uncover the underlying mechanisms by which genes influence disease processes and other biological phenomena.
Overview of scParser
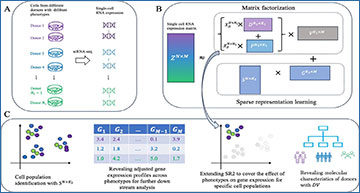
A The scRNA-seq data is obtained from biosamples from multiple biological conditions. Variations from biological conditions (e.g., donor and phenotypes) and technical artifacts may bring about batch effects in the scRNA-seq data. B scParser models the variation from donor and phenotype with matrix factorization and cellular variation with sparse representation learning. After cell population annotation, an extension of scParser enables revealing the effects of biological heterogeneous conditions (e.g., phenotypes) on different cell populations. C The output from scParser enables various interpretive data analyses, such as cell population annotation, connecting gene expression to biological conditions via biologically meaningful gene modules, revealing the heterogeneous effect of phenotypes on different cell populations via gene modules, and uncovering donors’ molecular characteristics
Enhancing Performance and Applicability
One of the key strengths of scParser is its ability to achieve superior performance in cell clustering compared to other leading methods. By accurately grouping cells based on their gene expression profiles, scParser provides clearer insights into how various biological processes are linked to disease development. Moreover, its broad applicability makes it a versatile tool for a wide range of research applications, from studying complex diseases to exploring fundamental cellular processes.
The Future of Data Analysis in Cellular Research
As scRNA-seq continues to evolve, the ability to integrate and analyze data effectively will be crucial for advancing our understanding of cellular biology. ScParser represents a significant step forward, offering a more nuanced view of how gene expression influences cell behavior and contributes to disease. With its scalable and insightful approach, scParser is set to become an invaluable resource for researchers aiming to decode the complexities of the cell and develop targeted therapies for various conditions.