Introduction: The Importance of RNA Modifications
RNA modifications, such as N6-methyladenosine (m6A) and N1-methyladenosine (m1A), play critical roles in regulating gene expression, RNA stability, and translation. These modifications add a layer of complexity to the transcriptome, influencing various cellular processes and responses. However, accurately detecting these modifications at a single-nucleotide level has been a challenging task, especially when dealing with highly modified sequences.
The Challenge with Traditional Detection Methods
Conventional techniques for detecting RNA modifications, such as mass spectrometry and sequencing-based approaches, often require prior knowledge of the sequence context or the use of enrichment strategies, which can limit their resolution and sensitivity. Moreover, these methods may struggle with densely modified regions, where the presence of multiple modifications can complicate accurate basecalling and detection.
Leveraging Nanopore Sequencing
Nanopore sequencing offers a unique advantage by allowing real-time analysis of nucleotide sequences, with the ability to directly detect modified bases. Despite its potential, the challenge has been to enhance the accuracy and reliability of detecting modifications without relying on sequence context.
Introducing Machine Learning for Enhanced Detection
In a recent study, researchers at University of Arizona have tackled this challenge by integrating machine learning into the nanopore sequencing workflow. Their approach utilizes incremental learning (IL) and anomaly detection (AD) to refine the process of basecalling and modification detection.
Step 1: Incremental Learning for Improved Basecalling
Incremental learning is employed to iteratively improve the basecalling accuracy of nanopore sequencing in sequences rich with modifications. By focusing on these biologically significant, modification-dense regions, the basecallers become better equipped to resolve complex sequences, enhancing the overall accuracy of the data.
Step 2: Anomaly Detection for Single-Nucleotide Resolution
Once the base sequence is accurately resolved, the next step is detecting the modifications themselves. The researchers used anomaly detection to identify modified nucleotides at a single-molecule, single-nucleotide level. This method allows for the detection of modifications without the need for prior sequence context, making it adaptable to a wide range of RNA molecules.
Adapt nanopore sequencing basecallers for modification detection
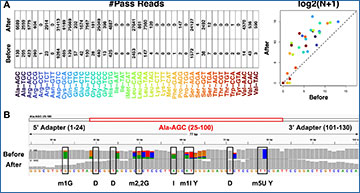
A Workflow overview. The move table records signal-nucleotide correspondences. C and M denote canonical and modified nucleotides, respectively. B, C Fully modified oligos cause basecalling failures, which can be explained by significantly shifted signals. Pass and fail denote nanopore sequencing reads that can and cannot be uniquely aligned to reference sequences, respectively. For each kmer instance, its signal shift was quantified against the canonical kmer model by a two-sided p-value. Raw p-values without adjustment yielded from the same kmer were concluded with an ecdf (empirical cumulative distribution function) curve. U.M. denotes unmodified sequencing reads. D, E Ameliorate basecalling failures with the incremental learning adaptation. Before and after denote pre and post-adaptation, respectively. F, H Per-read-per-site visualization of Ml-tags. Ml-tags represent modification scores. Without losing generality, 50 randomly selected full-length reads from contig 1 and 2 were visualized. G, I Quantify modification detection performance using confusion matrix statistics, including Balanced Accuracy (BA), Accuracy (ACC), True Positive Rate (TPR), and True Negative Rate (TNR). These statistics were calculated for each modification site at different Ml-tag thresholds, and visualized using density heatmaps. Positive and negative classes denote modified and unmodified nucleotides, respectively. Self and cross denote performance quantification on the same and the additional independent oligo ensembles, respectively.
Applications and Implications
The researchers validated their pipeline using control oligos and applied it to various biological samples, including densely modified yeast tRNAs, E. coli genomic DNA, and mammalian mRNAs. Notably, they achieved cross-species detection of m6A and simultaneous detection of m1A and m6A in human mRNAs.
This method represents a significant advancement in the field of epitranscriptomics, providing a powerful tool for exploring RNA modifications with high precision and minimal bias. By enabling the direct detection of modifications in their native sequence context, this approach has the potential to uncover new insights into RNA biology and the regulatory mechanisms governing gene expression.
A New Era for RNA Modification Research
The integration of machine learning with nanopore sequencing marks a transformative step in the study of RNA modifications. This approach not only enhances our ability to detect modifications at single-nucleotide resolution but also broadens the scope of research by eliminating the need for sequence context. As we continue to unravel the complexities of the epitranscriptome, such innovations will be crucial for advancing our understanding of RNA function and its role in health and disease.
Availability – The IL-AD workflow is available at: https://github.com/wangziyuan66/IL-AD.