Single-cell RNA sequencing (scRNA-seq) is a powerful technique that allows scientists to study individual cells in detail. This method is particularly useful because it can reveal how different cells in the same tissue may have distinct functions. However, analyzing scRNA-seq data is challenging because it involves working with a large number of genes and relatively few cells. Moreover, not all detected genes contribute to the specific functions of each cell type, which makes it difficult to identify the most important genes for understanding how cells operate.
Introducing COMSE
Researchers at Tsinghua University have developed a new tool called COMSE (Community-based Feature Selection for Single-cell RNA-seq data), designed to make analyzing scRNA-seq data more effective. COMSE works by selecting the most informative genes from the data, which are the ones that play key roles in the biological processes of different cell types.
One of the key features of COMSE is that it can identify different “substates” within cells. A substate refers to a specific condition or phase that a cell might be in at a particular time. For example, COMSE can distinguish between cells that are in different stages of the cell cycle—a process that involves cell growth and division. This ability to pinpoint such subtle differences is crucial for understanding how cells function and interact within tissues.
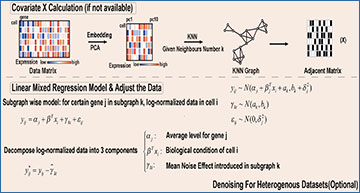
An overview of the COMSE method for selecting informative genes from single-cell RNA-seq data
A The log-normalized gene expression profile of each cell is projected into a low-dimensional latent space using principal component analysis (PCA). The low-dimensional representation is then used to construct a gene similarity graph with the K-nearest neighbors (KNN) algorithm. Gene graph is partitioned into several subgraphs by Louvain algorithms for community detection. B When sample covariates lacking, covariate matrix for each cell were estimated through KNN in low-dimension PCA space with given neighbor number. Then linear mixed regression model was applied to estimate and remove noise from data within each gene subgraph. C An unsupervised feature selection technique based on the Laplacian score is applied to choose HIGs in each subgraph
How COMSE Outperforms Other Methods
COMSE has been tested on both real and simulated scRNA-seq datasets, and it has shown impressive results. Even when the data had high dropout rates (meaning that some gene information was missing), COMSE was still able to accurately cluster cells based on their similarities. This means that COMSE is particularly good at grouping cells into categories that reflect their true biological states, even when the data is incomplete or noisy.
Additionally, COMSE can identify and correct for “batch effects.” Batch effects occur when differences in data arise not because of biological differences, but because of variations in how the data was collected, such as using different sequencing protocols. By detecting communities of genes that are associated with these technical differences, COMSE helps ensure that the analysis reflects true biological signals rather than noise.
Why COMSE is Important
The development of COMSE represents a significant advancement in scRNA-seq analysis. By focusing on the most informative genes, COMSE allows researchers to more accurately identify cell subtypes and understand how cells differ from one another. This can lead to better insights into how tissues function, how diseases develop, and how different cells might respond to treatments.
Moreover, COMSE’s ability to handle batch effects makes it a valuable tool for integrating data from different sources, which is increasingly important as more and more scRNA-seq datasets become available. Beyond single-cell analysis, COMSE also performs well in analyzing bulk RNA-seq data, making it a versatile tool for various types of genomic studies.
In summary, COMSE is a powerful, unsupervised framework that enhances the analysis of scRNA-seq data by selecting the most relevant genes, improving the identification of cell subtypes, and correcting for technical biases. This makes it an important tool for researchers working to understand the complexities of cellular function and disease.
Availability – The source code for COMSE is implemented in R and can be found at https://github.com/Lan-lab/COMSE.